No results found
We couldn't find anything using that term, please try searching for something else.

Tailoring van der Waals dispersion interactions with external electric charges
Intermolecular interactions from quantum Drude oscillatorsElectrons in systems with finite band gaps,such as organic molecules,nonmetallic solids,and
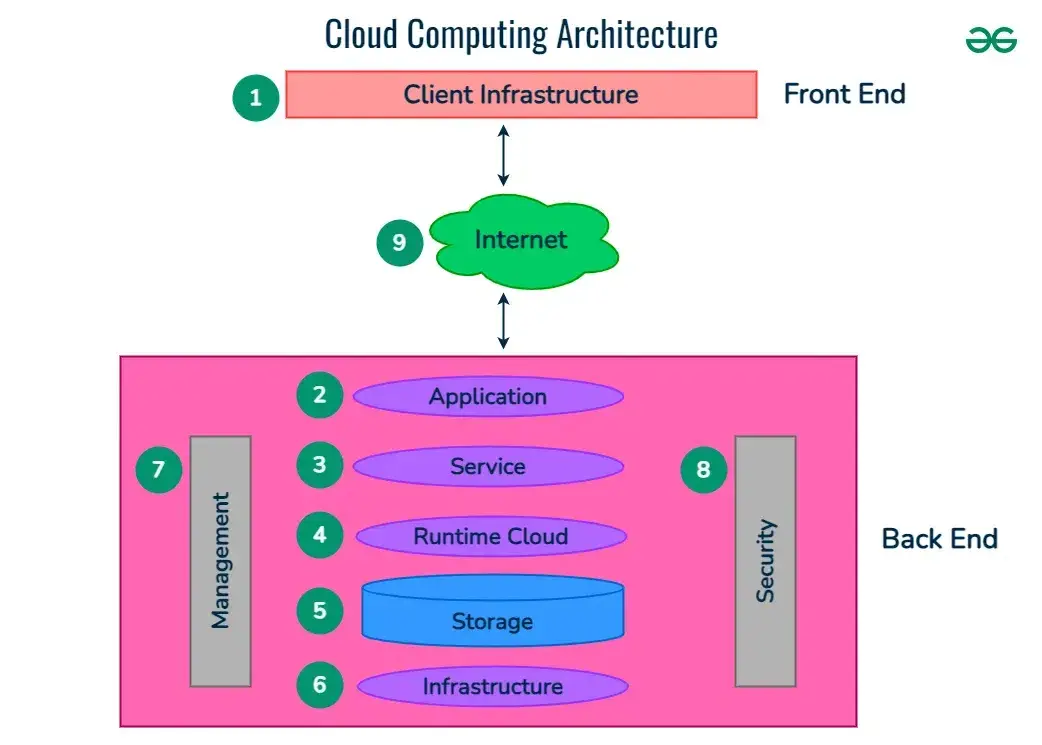



Intermolecular interactions from quantum Drude oscillators
Electrons in systems with finite band gaps,such as organic molecules,nonmetallic solids,and nanostructures,are well described by a localized representation. Therefore,collective charge density fluctuations in these systems arise from the dynamically correlated motions of local multipole excitations. Accordingly,we treat the response of valence electrons of a given molecule as that of a set of interacting atomic response functions. Quantum-mechanical parameterization of the valence electronic response in terms of coupled atomic fluctuations is done efficiently andaccurately within the quantum Drude oscillator (QDO) approach22,23,24,25,26,27,28,29,30,31. The QDO model replaces oscillations of the electron cloud on each atom with an effective quantum harmonic oscillator,characterized by a set of three effective parameters: mass,frequency,and charge (μ,ω,q)24,25,26.
The couple Drude oscillator model has been show to yield a quantitatively accurate description of many – body dispersion interaction in the dipole limit29,31,32,33,which makes it a promising approach for higher multipole generalization andcoupling to external electric fields26,28,34,35.
The hamiltonian of a system of n QDOs interacting via Coulomb forces between themselves andM point charges δj,placed at \(\widetilde {\it{r}}_j\),is given by:
$$\begin{array}{*{20}{l}} h \hfill & = \hfill & {\mathop {\sum}\limits_i^n {\kern 1pt} h_{0i} + \mathop {\sum}\limits_i^n {\kern 1pt} \mathop {\sum}\limits_j^M {\kern 1pt} \delta _jq_i\left( {\frac{1}{{\left| {{\it{r}}_i \hskip 1pt-\hskip 1pt \widetilde {\it{r}}_j} \right|}} – \frac{1}{{\left| {{\it{r}}_i \hskip 1pt-\hskip 1pt \widetilde {\it{r}}_j} \right|}}} \right)} \hfill \\ {} \hfill & {} \hfill & { + \hskip 1pt\frac{1}{2}\mathop {\sum}\limits_{i \ne i\prime }^n {\kern 1pt} q_iq_{i\prime }\left( {\frac{1}{{\left| {{\it{r}}_i \hskip 1pt-\hskip 1pt {\it{r}}_{i\prime }} \right|}} + \frac{1}{{\left| {{\it{r}}_i \hskip 1pt-\hskip 1pt {\it{r}}_{i\prime }} \right|}}} \right.} \hfill \\ {} \hfill & {} \hfill & {\left. { -\hskip 1pt \frac{1}{{\left| {{\it{r}}_i \hskip 1pt-\hskip 1pt {\it{r}}_{i\prime }} \right|}} – \frac{1}{{\left| {{\it{r}}_i \hskip 1pt-\hskip 1pt {\it{r}}_{i\prime }} \right|}}} \right),} \hfill \end{array}$$
(1)
whereh0i = \(- \frac{{\hbar ^2}}{{2\mu _i}}\nabla _{{\it{r}}_i}^2 + \frac{1}{2}\mu _i\omega _i^2\left( {{\it{r}}_i – {\it{r}}_i} \right)^2\) is the unperturbed QDO hamiltonian,assuming fixed oscillation center position ri andri is a position of the Drude particle. Details of the unperturbed QDO solution may be found in the Supplementary note 1. Equation (1) is similar to the conventional molecular hamiltonian,however the full electronic-nuclear system is replaced by Drude quasiparticles,placed on each atom. Owing to its quantum nature,the QDO represents a spatial distribution of charge,which could be modified by other QDOs or external fields,giving this model the ability to capture complex electronic response effects. Owing to the full Coulomb coupling between charges,the exact quantum-mechanical solution of the QDO hamiltonian in Eq. (1) contains all multipoles andmany-body effects to all orders of perturbation theory26. This makes the QDO hamiltonian an optimal starting point to develop approximations. For example,within classical mechanics one would recover polarizable force fields36,37,38,whereas within the quantum-mechanical dipole approximation one obtains the previously developed many-body dispersion (MbD) model29.
here,we study the effect of an electric field induced by an external point charge on dispersion interactions between QDOs in the linear response regime. We initially consider a system made of two QDOs,A andb,and a point charge δ,placed at distances \(\tilde r_{A/b}\) from them (see Fig. 1) . The quantum-mechanical Drude model allows to describe electrostatics,induction,and dispersion effects in isolation andinduced by external charges. The approach presented here could be generalized to a set of external charges andQDOs in a straightforward manner.
Fig. 1
Model for field-induced dispersion. A system made of two QDOs with distance r between them anda point charge δ placed at a distance \(\tilde r_A\) and\(\tilde r_b\) from the oscillator centers A andb, respectively. QDOs are sketched by blue circles with red oscillation center andyellow oscillating Drude quasiparticle. The external charge is marked by a green dot
The last two term in Eq . ( 1 ) could be treat as perturbationh′ = hA + hb + hAb of h0,which are caused by the interaction of each QDO with an external point charge hA/b,and the interaction between QDOs,hAb. henceforth,we use the indices A andb to mark variable relate to different oscillator . In term of the spherical multipole tensorQlm (of order l,−l ≤ m ≤ l),the interaction of test charge δ with a negatively charged Drude quasiparticle andpositively charged oscillation center is expressed in a compact form3,26: hA/b = \(- \delta \mathop {\sum}\nolimits_{l = 1}^\infty {\kern 1pt} \mathop {\sum}\nolimits_{m = – l}^{m = l} {\kern 1pt} Q_{lm}^{A/b}{\mathrm{/}}\tilde r_{A/b}^{l + 1}\).
The interaction between two QDOs at a distance r between them is given by the multipole interaction tensor \(T_{lm;l\prime m\prime }^{Ab}\),which depends on this distance andrelative orientation3,26,39,40,41,42,43: hAb = \(\mathop {\sum}\nolimits_{l,l\prime = 0}^\infty \mathop {\sum}\nolimits_{m,m\prime } {\kern 1pt} Q_{lm}^AT_{lm;l\prime m\prime }^{Ab}Q_{l\prime m\prime }^b\). The response to an external electric field produced by a charge is given by the polarizabilities αlm,l′m′,defined in second-order rayleigh-Schrödinger perturbation theory3,26. herein,we consider input atomic polarizabilities to be isotropic αlm,l′m′ = \(\alpha _ l\delta _ { l , l\prime } \delta _ { m , m\prime } \ ),wherel define the multipole order (l = 1 corresponds to the dipole,l = 2 to quadrupole,etc.) andαl is give by the analytical expressionαl = \(\left ( { \frac{{q^2}}{{\mu \omega ^2 } } } \right)\left [ { \frac{{(2l – 1)!!}}{l } } \right]\left ( { \frac{\hbar } { { 2{\kern 1pt } \mu \omega } } } \right)^{l – 1}\ )26. This is an excellent approximation within the employed atomic Tkatchenko–Scheffler model (see below andin ref. 44) .
Analytic expression for the field–induced dispersion energy
building the perturbation theory on eigenvectors of h0 andtreating h′ as a perturbation,Martyna andco-authors developed a Jastrow-type diagrammatic technique26,45,which proves to be a very useful tool for understanding coupled QDOs. In brief,diagrammatic rules are the following: the yellow blocks are associated with the electrostatic multipole interaction between two QDOs andtheir number indicates successive orders of perturbation expansion; connectors,coming out of one end of a yellow bar,show the multipole order. The asymptotic interaction power laws are given by combination of r and\(\tilde r\) andcan be obtained by analyzing separate parts of diagrams: yellow bar gives r′−1 decay andconnector contributes as r′−2,where\(r\prime \in \left\{ {r,\tilde r} \right\}\). For the detailed discussion of the diagrammatic principle we is refer refer the reader to ref .26 andreferences therein. Several representative diagrams for two (three) QDOs are shown in Fig. 2. Within this framework,dispersion terms arise from the bubble-like closed diagrams (Fig. 2c,f,h,i) andthe open ends indicate the polarization terms (Fig. 2b,d,e),which arise from interaction with external fields.
fig . 2
Diagrammatic expansion of interacting quantum Drude oscillators. representative low-order interaction energy diagrams for coupled QDOs,associated with the following expansion terms: a bare Coulomb electrostatic interaction ;b dipole polarization; c conventional vdW dipole–dipole dispersion; d quadrupole polarization ;e many-body dipole polarization; f dipole–quadrupole dispersion; g dipole – quadrupole electrostatic interaction ;h three-body dipole–dipole-dipole dispersion; i charge-induced pairwise dipole–quadrupole dispersion (called FID in this paper) . QDOs are sketched by blue circles with red oscillation center andthe external charge is shown with a green dot,polarization is indicated with elongation of the inner circle
In this work,we focus on the leading dispersion term modified by an external charge δ,corresponding to the dipole–quadrupole polarization–dispersion interaction (Fig. 2i),which appears in the third order of perturbation theory. This diagram arises from the dispersion interaction between two QDOs,one being in a dipolar state,the other in an excited quadrupolar state owing to polarization via the external charge. The diagram on Fig. 2i is not symmetric,as it relates to the polarization of one out of two interacting QDOs. however,the total dipole–quadrupole charge-induced dispersion (FID–(DQ)) energy should be completed with another diagram that relates to the quadrupole polarization of the second QDO. The sum of these two diagrams will be referred to as EFID in this study.
This first non-trivial FID diagram,shown on Fig. 2i,translates into the analytic form as follows (details of the derivation andgeneral form of the dispersion terms are provided in Supplementary note 1 along with discussion of Fig. 2g):
$${\hskip-36pt}E_{{\mathrm{FID}}}^A = – \frac{\delta }{2}\frac{{\alpha _1^b\alpha _2^A\omega _A\omega _b}}{{\left( {2\omega _A + \omega _b} \right)\left( {\omega _A + \omega _b} \right)}}\frac{1}{{\tilde r_A^2}} {\hskip36pt}\\ \times \mathop {\sum}\limits_{m_A,m_b} {\kern 1pt} T_{2,- m_A;1,- m_b}T_{1,m_A;1,m_b}\sqrt {4 – m_A^2}$$
( 2 )
Equation (2) depends quadratically on \(\tilde r_A^{ – 1}\) andlinearly on δ,reflecting that only one QDO is polarized by the external charge. This dispersion term scales as r−7 with distance between two oscillators,which makes it noticeable compared with the conventional dipole–dipole dispersion interaction decaying as r−6. Also we note that the FID energy is a purely quantum effect,as α2 ∝ ℏ vanish in the classical limit for a QDO26. A similar effect is expected in case of other non-uniform electric fields,for example that induced by a finite-size dipole. FID would scale as \(\tilde r^{ – 9}\) with the distance to the dipole,making dispersion induced by a point charge a leading-order effect.
In this work,we model each atom by a separate QDO andparametrize it with a Tkatchenko–Scheffler (TS)-like approach46,extended to the quadrupole polarizability in the following way (see Supplementary note 1 for details): \(\alpha _2^{{\mathrm{eff}}}\) = \(\alpha _2^{{\mathrm{free}}}\left( {\frac{{V_h^{{\mathrm{eff}}}}}{{V_h^{{\mathrm{free}}}}}} \right)^2\). This approach takes into account local electronic environment by weighting the free-atom quadrupole polarizability \(\alpha _2^{{\mathrm{free}}}\)47,48 with the ratio between free andeffective hirshfeld volumes. The angular dependence of dipole–quadrupole FID is expressed by the sum over angular momentum indices m of the interaction function product. It takes the simplest form when two identical QDOs \(\alpha _l^A = \alpha _l^b = \alpha _l\) lie on the same z axis with the same distance to the external charge \(\tilde r_A = \tilde r_b = \tilde r\). In this case,the frequency dependence is canceled out andthe sum of two QDOs FID contribution \(E_{{\mathrm{FID}}}^A + E_{{\mathrm{FID}}}^b\),given by Eq. (2),simplifies to:
$$E_{{\mathrm{FID}}} = – 3\delta \alpha _2\alpha _1\frac{1}{{\tilde r^2}}\frac{1}{{r^7}}.$$
(3)
We note that a positive sign of the external charge δ > 0 corresponds to a negative energy contribution EFID < 0. An intuitive mechanism behind this charge dependence is the following: as positive δ attracts the interacting pair of Drude particles,it polarizes both their orbitals in the same direction toward the external charge,which leads to stabilization. Similar arguments are valid for the inverse effect in the field of a negative charge. These findings could be explained in terms of the hellmann–Feynman theorem as well. External charge populates excited states of the QDO system,leading to anisotropic delocalization of the electron cloud,which gives rise to Feynman forces49. Therefore,alternatively one can derive the FID contribution in terms of high-order hyperpolarizabilities,similarly to the Feynman dipole approach described in ref.50. We note,however,that the FID contribution is present already in the linear response regime.
Along with the dispersion term,described by Eq. (2),the total energy of the system contains classical electrostatic interaction terms,which might be important for polar molecules. however,these terms do not scale with molecular polarizability,hence dispersion interactions remain dominant for large molecules. Furthermore,electrostatic interactions are already included in force fields andDFT calculations employing semilocal functionals. In contrast,to the best of our knowledge FID terms have so far not been included in any atomistic simulation. next,we will show that FID contributes significantly to the binding energies of relatively small molecules andits importance grows with molecular size andpolarizability.
Field–induced dispersion interaction between small molecular dimers
before presenting the importance of FID effects in biological systems,we first discuss the FID contribution to the correlation part of the binding energy between small molecular dimers andbenchmark our analytic results (see Eq. (2)) with direct ab initio calculations. Ab initio calculations were carried out using the rPA method51,52 converged at a complete one-electron basis set limit (see Methods section for details),and these results serve as an accurate reference for comparison with analytic FID values. The rPA approach computes the correlation energy to infinite order in perturbation theory andtreats the influence of an external charge on excited states of molecules,hence being an appropriate reference for FID.
We start by benchmarking the FID effect on a molecular cyclopentane dimer system. Figure 3 shows EFID as a function of the cyclopentane dimer center of mass separation,whereas keeping fixed the distance from each QDO to the external charge,\(\tilde r_A = \tilde r_b = 5\) Å. The interaction energy computed with the analytic formula in Eq. (2) is in excellent agreement with reference rPA calculations. In addition,both rPA andanalytic FID formula are essentially antisymmetric with respect to the sign of the external charge. In contrast,all popular methods for vdW interactions in DFT are unable to capture the FID effect,as the direct change in the electron density by external charge is negligible (identical conclusions hold for vdW methods such as D3,vdW-DF,XDM,TS,and MbD29,46,53,54,55,56,57) . Molecular orbitals employed in second-order Møller–Plesset perturbation theory (MP2) framework are more responsive to electric fields than the ground-state electron density. hence,MP2 correlation energy is affected by external charges. however,MP2-binding energies in this case arise from hyperpolarization effects (not linear polarization as in rPA) andare not antisymmetric with respect to δ → −δ,having a smaller magnitude compared to the reference rPA calculations (also see Table 1) . This is particularly significant for practically relevant distances,when the external charge is close to the molecule.
Fig. 3
Field-induced dispersion effect in cyclopentane dimer. FID as a function of intermolecular distance r in Å,calculated at a fixed distance to the external unit charge \(\tilde r = 5\) Å for the cyclopentane dimer (geometry shown as inset) . The analytical result is shown in blue. The reference rPA@PbE0 calculations are marked with a red curve. The effect of the charge in conventional vdW methods such as MbD@PbE0 (purple) is negligible. The effect of the charge on MP2@hF correlation energies (dashed green) is visible,however having a different origin than FID. Upper positive halfplane corresponds to the negative sign of the external charge δ = −1,and negative—to the positive sign δ = 1
Extending our study to larger systems,we tested the magnitude of FID effect on molecular dimers from the S66 benchmark database of non-covalent interactions58 (see Table 1) . We consider dimer geometries at equilibrium separation58. We chose the position of the external charge such that the electric field at the center of mass of the dimer reaches 109– 1010 V/m,by placing an external charge at 3 or 5 Å. This range of distances reflects realistic environments present in biological systems. FID calculations are summarized in Table 1,wherethe following notation is used. The molecular dimer correlation binding energy in the presence of an external charge δ is define as :\(E_c^{{\mathrm{bind}}}(\delta )\) = \(E_c^{Ab}(\delta ) – E_c^A(\delta ) – E_c^b(\delta )\),where\(E_c^{Ab}\) is a correlation energy of the molecular dimer,and \(E_c^{A/b}\) are the correlation energies of the separate moieties A andb. The effect of the external charge on correlation energy is given by the difference: \({\mathrm{\Delta }}E_c^{{\mathrm{bind}}}(\delta )\) = \(E_c^{{\mathrm{bind}}}(\delta ) – E_c^{{\mathrm{bind}}}(\delta = 0)\ ). In order to separate the vdW dispersion contribution in Eq. (2) from other terms of electrostatic-induction origin (see Supplementary Fig. 1) we use the fact that FID changes sign with δ anddefine the following linear combination: \(W_c^{{\mathrm{bind}}}(\delta ) \ ) = \({\textstyle{1 \over 2}}\left [ { e_c^{{\mathrm{bind } } } ( – \delta ) – E_c^{{\mathrm{bind}}}(\delta ) } \right]\ ). Overall,our analytic formula for FID yields an excellent agreement with full rPA calculations. Largest deviations amount to 30% andstem mainly from the fact that our formula is the leading-order term,whereas rPA includes contributions up to infinite order. Our model can be generalized to infinite order in a straightforward way as a potential future work.
There are several noteworthy observations that can be drawn from Table 1. First,the FID energy can reach up to 35% (47.4 meV) of the total binding energy for the cyclopentane dimer. FID also contributes substantially in benzene,n-methylacetamide andpyrazine dimers. Second,it is clear that existing methods for vdW dispersion energy in DFT29,46,53,54,55,56,57 are unable to describe FID because they do not have any mechanism to couple to external electric fields. In contrast,MP2 correlation energies are affected by the external charge due to orbital polarization via the external charge. however this effect is smaller than reference rPA@PbE0 results andnon-symmetric with respect to the change of the external charge sign (compare second column in Table 1 with fourth andfifth columns) .
For non-polar molecules (i.e.,ammonia,cyclopentane,n-methylacetamide),FID energies are larger or comparable to the electrostatic contribution to the binding energy in the presence of an external charge (see Supplementary Table 1) . In general,electrostatics will be dominant for polar molecules (i.e.,water dimer in the last row of Table 1) . however,the sign of the FID contribution is proportional to −sign(δ) in contrast to the charge-induced electrostatic term,which depends on the relative orientation of the molecular dipole. Consequently,in many practical situations,we expect an interplay between electrostatic energies andFID andtuning this delicate balance paves a novel way to control structure anddynamics via externally applied electric fields.
Field-induced dispersion interaction in biological systems
In this section,we discuss the importance of FID in biological systems,namely in ionic channel models (Fig. 4) andamino-acid dimers (Fig. 5),both of which are key to biomolecular function. Amino acids serve as building blocks for proteins,which are in turn essential components of living cells. Amino acids are responsible for ion channel formation that serve for selective ion transport through the cell membrane7. Ion mobility can be controlled by external stimuli such as voltage,mechanical stress,protein phosphorylation,ligand binding,or changes in ph7. The last two gating mechanisms occur in the presence of external charges,and here we show that FID energies may contribute substantially to these processes.
fig . 4
Field-induced dispersion effect in the selectivity filter of Kcv ion channel. a Cross section of selectivity filter of a representative ba2 + ion channel59 andzoomed S4 region with three threonine andone serine molecule (baT3S1); b Absolute value of FID \(W_c^{{\mathrm{bind}}}(\delta = + 2)\) (in meV) obtained from numerical rPA correlations (red line) andanalytic Eq. (2) (blue line) compared with the MbD energy (yellow bars) for the ba2 + complex with four amino-acid ligands; c Substitutional MbD energies (shown in yellow) andsubstitutional FID energy contribution (blue) for the same ion complexes
Fig. 5
Field-induced dispersion in amino-acid dimers. FID \(W_c^{{\mathrm{bind}}}(\delta = 1)\) energy (in meV) obtained from rPA calculations (red line) andanalytic Eq. (2) (blue line) compared with the MbD binding energy (yellow bars) . The geometry of phenylalanine dimer anda point charge (in pink) is shown in the inset
We start with a set of amino-acid dimers at equilibrium separation with their geometries optimized using the DFT + MbD29 method in the absence of external fields. Subsequently,a point charge was added at 3.5 Å distance from the center of mass of the dimer andthe FID energy was determined,by carrying out rPA calculations (see Supplementary Table 2 for details) . The results are summarized in Fig. 5,wherewe show FID contributions obtained from rPA correlations andthe analytical formula in Eq. (2) . In order to assess the significance of the FID contribution,we compare it with the dispersion binding energy obtained with MbD for the amino-acid dimer in absence of any field. remarkably,FID contribution reaches up to 35% of the dipole–dipole dispersion binding energy,varying in the range from 3 to 15% of the total binding energy. hence,we conclude that FID energies for biological systems can substantially exceed 1 kcal/mol—the minimum desired level of accuracy for atomistic modeling.
As a final example we consider the selectivity filter region of the Kcv ion channel59 anddispersion interactions therein. We focus on one of the preferred binding sites—S4 (shown in Fig. 4a),that is typically composed of four threonine residues that provide four backbone carbonyl oxygens andfour side-chain hydroxyl oxygens for ion coordination59,60,61. S4 site plays a special role in the ion gating mechanism,as ba2 + ions can bind to this site as well,blocking K+ permeation60,62. besides ion substitution,sequence decoding shows that within the channel a threonine residue could be replaced by serine (identical to threonine except for one missing methyl group)60 (see Fig. 4a) . Site-directed mutagenesis experiments suggest that threonine-to-serine (T → S) substitution in the S4 sites reduces the channel susceptibility to ba2 + andits overall opening probability61. The key role of methyl groups for understanding the polarization andas a consequence vdW interactions in Kcv channels as well as dramatic ion affinities reduction during T → S substitutions was already suggested61. This leads us to the question of whether an inhomogeneous electric field could have considerable effect on dispersion interactions andbinding in Kcv channel beyond direct electron density polarization. To address this question,we first compare FID energies to the MbD-binding energy of ba2 + ( see Supplementary Fig . 2 for K+ andna+ ions) for isolated S4 site geometries,composed of threonine or serine residues (Fig. 4b) . As the ions are positively charged,FID stabilizes dispersion energy with a relative contribution ranging from 10% in the case of K+ to 30% for ba2 + complex,having a considerable effect in the total binding energy (total energies are shown in the Supplementary Tables 3–5) .
To better understand the role of FID andits contribution to the vdW dispersion energy,we now consider substitution energies for the methylation process in ionic channels. The substitution energy is calculated as the energy difference between the left andright hand side of the following reaction:
$ $ AT_4 is \hskip + ns \hskip 1pt\rightleftharpoons \hskip 1ptAT_{4^\_n}S_n + nT,$$
(4)
wheren is the unit increment of threonine vs. serine substitutions,A represent one of the k+,na+,ba2 + ions andnT or nS are amino acids in the gas phase. These substitutions result in minor geometry changes,so we used geometries reported in ref.61,modeling an ion by a point charge. Corresponding substitution binding energies of the ba2 + complex,wherethreonine groups were systematically substituted by serine groups,are shown in Fig. 4c (see Supplementary Tables 3–5 for total binding energies) . Substitutional energies are also largely affected by FID. One T → S substitution (n = 1) almost doubles FID stabilization of the dispersion energy andreaches −35 meV in the case of complete T → S substitution. The main finding here is that FID enhances the methylation stabilization effect. Overall,our results provide compelling evidence that FID is an essential energy contribution to biological systems in the presence of external electric fields.
Figure 4b also indicates the need for further theoretical developments of the FID model. namely,the largest deviation between reference rPA calculations andanalytical FID model occurs for the most polarizable baT4 system andsuggests that higher-order polarization–dispersion terms beyond that shown in Fig. 2i must be included to achieve a quantitative treatment of FID effects.